
几种基于核酸的PROTAC简介
近年来,核酸药物发展迅猛,市场需求不断增加,上市审批迅速,涵盖心血管与代谢疾病、肝病、肿瘤等多个领域。截至目前,全球已有10余种核酸药物获批上市,不少核酸药物正处于临床试验阶段。核酸药物有望成为继小分子药物、抗体药物之后的第三类药物(图1)。

越来越多经批准的核酸疗法证明了通过靶向体内致病基因来治疗疾病的潜力。通常,常规治疗只产生短期治疗效果,因为它们针对的是蛋白质而不是疾病的根源,而核酸药物则直接作用于致病的靶基因或靶mRNA,在基因水平上发挥治疗疾病的作用。核酸药物包括ASO、siRNA、Aptamer、miRNA、mRNA、saRNA、sgRNA、U1 snRNA等。核酸药物具有治疗效率高、毒性低、特异性强、应用领域广泛等优点,显示出其重要的医学价值、生物科学等领域。
PROTAC(蛋白水解靶向嵌合体)是一种利用泛素-蛋白酶体系统(UPS)降解靶蛋白的药物开发技术。从结构上看, PROTAC由三部分组成:E3泛素连接酶配体和靶蛋白配体,两个活性配体通过专门设计的“Linker”结构连接在一起,形成三元复合物。 PROTAC的靶蛋白配体与靶蛋白结合,E3泛素连接酶配体与胞内E3泛素连接酶的底物结合区结合,从而通过泛素化靶蛋白,将靶蛋白“拉”到E3泛素连接酶上,使 UPS 系统能够降解目标蛋白(图 2)。

过去20年来,研究人员基于肽和小分子设计了各种形式的PROTAC。然而,基于肽的PROTAC存在活性低和免疫原性等问题,极大地限制了其临床医学应用。与多肽PROTAC相比,小分子PROTAC体积更小,更容易被人体吸收,成药性更好,因此小分子PROTAC仍然是主流。随着科学技术的发展和进步,一些新型的PROTAC不断出现,基于核酸的PROTAC应运而生。
RNA-PROTAC
RNA 结合蛋白 (RBP) 的功能缺陷是许多疾病的根源,而用常规药物靶向 RBP 已被证明是困难的。 RBP 以动态、协调和序列选择性的方式与 RNA 结合,形成核糖核蛋白 (RNP) 复合物,在 RNA 依赖性中发挥关键作用。某些疾病是由 RBP 的基因变化引起的,影响了 RBP 与 RNA 的结合。
RNA-PROTAC 的新型嵌合结构来靶向 RBP。 2021年,Jonathan Hall研究小组提出基于RNA的PROTACs的设计理念,作者成功构建了靶向 RBP(RNA 结合蛋白)的 RNA-PROTAC。使用小 RNA 模拟物作为靶向基团,它们可以特异性结合 RBP RNA 结合位点。 PROTAC 泛素化 RBP,然后通过泛素蛋白酶体系统降解它们。作者对两种 RBP(干细胞因子 LIN28 和剪接因子 RBFOX1)的降解进行了概念验证,并展示了它们在癌细胞系中的用途。 RNA-PROTACs靶向Lin28蛋白,这是一种干细胞因子和癌蛋白,是多种疾病的潜在药物靶点,具有很高的研究价值。

TF-PROTAC
转录因子(TF)是治疗包括癌症在内的疾病的一类重要治疗靶点。由于TFS缺乏激酶或其他酶中常见的活性或变构位点,传统的小分子抑制剂很难与其结合。因此,转录因子一度被认为是“不可成药”的靶点,存在难以逾越的技术瓶颈。哈佛大学Wenyi Wei教授和西奈山伊坎医学院Jian Jin教授报道了一种基于寡核苷酸链的“ TF-PROTAC “,它由DNA寡核苷酸和E3配体通过点击反应连接而成(图4),并能选择性地降解致病性TF。 TF-PROTAC 的选择性取决于所使用的 DNA 寡核苷酸。
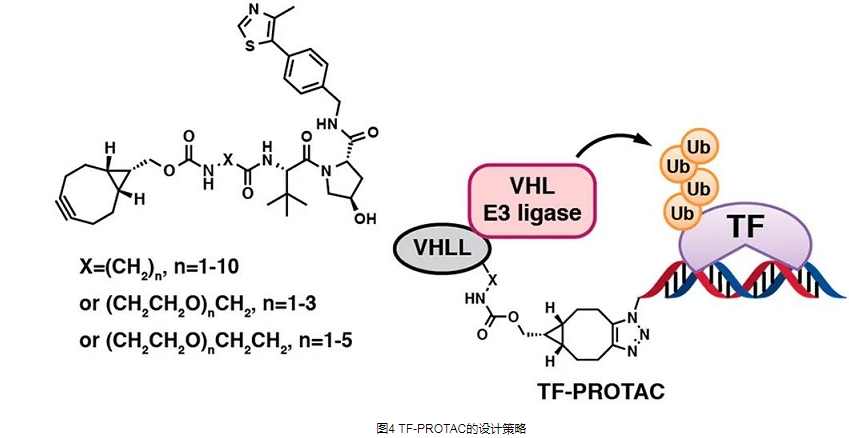
作者成功开发了两个系列的基于VHL的TF-PROTAC:NF-κB-PROTAC(dNF-κB)和E2F-PROTAC(dE2F),分别有效降解细胞内的内源性p65和E2F1蛋白,并表现出优异的抗-细胞增殖作用(图5)。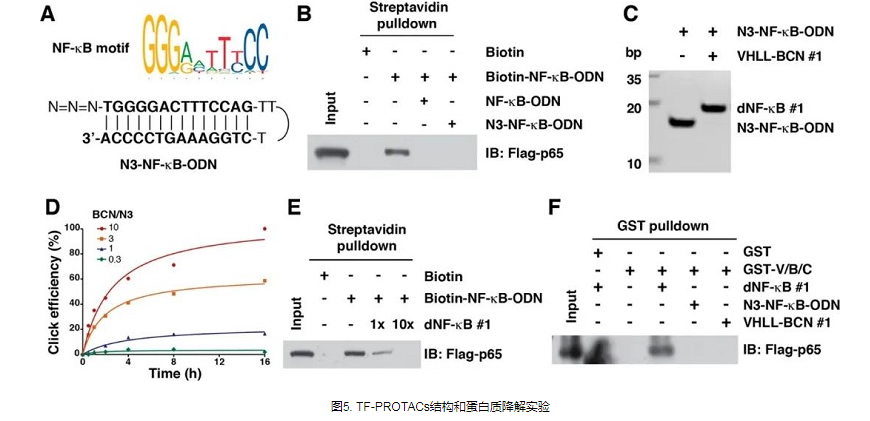
2021 年,Crews 团队还报道了靶向转录因子 (TF) 的寡核苷酸 PROTAC:oligoTRAFTAC(图 6)。OligoTRAFTAC由与 TF 和 E3 泛素连接酶配体结合的寡核苷酸链组成。
蛋白质印迹实验表明,oligoTRAFTAC 成功降解了两种致癌转录因子:c-Myc 和 brachyury。此外,作者发现oligoTRAFTACs可以成功降解脊索瘤细胞系的brachyury,并且在随后的体内斑马鱼模型实验中也表现出良好的降解活性(图7)。
基于适配体的PROTAC
适体是寡核苷酸(DNA 或 RNA)序列。寡核苷酸片段通常通过指数富集配体系统进化(SELEX)体外筛选从核酸文库中获得。核酸接头因其可以与多种目标物质以高特异性和选择性结合而被广泛应用。 2021年,谭卫红课题组基于核酸适配体AS1411:ZL216设计了PROTAC(图8)。

AS1411可以特异性靶向肿瘤细胞中高表达的核仁素受体,核仁素受体与其配体结合后被内化。作者通过体外实验证明PROTAC具有较高的水溶性和血清稳定性。此外,作者发现ZL216促进乳腺癌细胞中核仁素受体-ZL216-VHL三元复合物的形成,并在体外和体内有效诱导核仁素受体降解。随后的细胞增殖和迁移实验表明,ZL216还抑制乳腺癌细胞的增殖和迁移(图9)。

适体-PROTAC 缀合物 (APC)
PROTAC是一种很有前景的靶向蛋白质降解策略。作为一种有效的靶向蛋白质降解方法,PROTACs在催化性能、高选择性、克服耐药性、有效阻断非成药靶点等方面显着优于传统小分子药物。但PROTAC一般具有高分子量和高疏水性,其理化性质很大程度上超过了“五法则”(RO5)。因此,传统PROTAC的药物开发往往受到其细胞膜通透性差、药代动力学(PK)特性差以及缺乏肿瘤特异性靶向的限制。
为此,盛春泉课题组提出了Aptamer-PROTAC Conjugates(APCs)的设计理念。 APC 是通过可裂解的连接链将 PROTAC 靶向 BET 蛋白与适体 AS1411 (AS) 偶联而获得的(图 10)。其中,核酸适体AS1411可以选择性靶向肿瘤细胞表面高表达的核仁素受体。 AS本身对核仁素受体过表达的肿瘤具有良好的抑制活性,目前正在II期临床试验中进行评估。肿瘤细胞中谷胱甘肽含量丰富,因此连接链选择可被谷胱甘肽(GSH)裂解的二硫键,可选择性响应肿瘤微环境,在连接链裂解后释放活性BET降解剂。
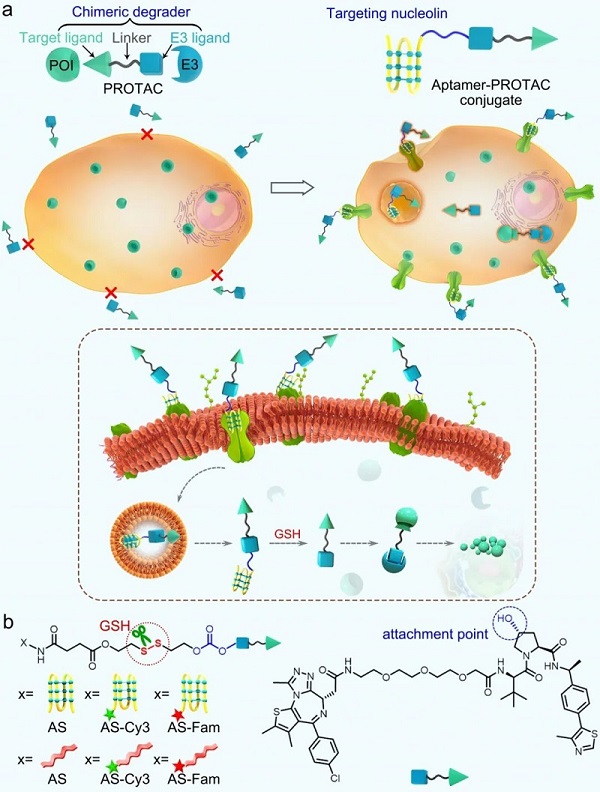
图 10. APC 的设计策略
与未修饰的BET PROTAC(PRO)相比,APC缀合物(APR)在McF-7细胞的小鼠异种移植模型中表现出改善的肿瘤靶向能力,从而增强BET蛋白的体内降解和抗肿瘤效力。因此,APC策略为肿瘤特异性靶向PROTAC的开发提供了新的设计思路。
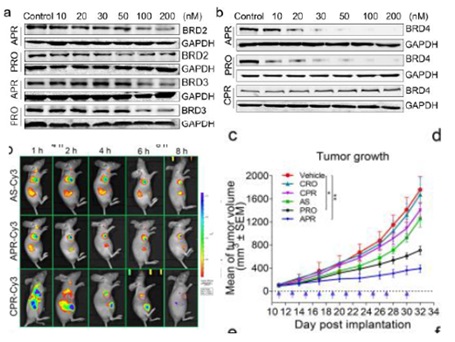
图11 蛋白质降解实验及体内成像、抗肿瘤实验
适配体PROTACs Conjugates (APCs)的发展自提出以来可谓“惊天动地”。与传统PROTAC相比,基于核酸的PROTAC提高了传统小分子PROTAC的靶向性,在提高水溶性、膜通透性、肿瘤靶向性等方面发挥着重要作用。由于核酸药物在体内易被核酸酶水解,半衰期短,极大限制了其在生物医学中的应用。未来的研究方向应集中于提高核酸药物的稳定性、延长半衰期、改善药代动力学性质、解决核酸药物递送问题。
作为专业的PEG衍生物供应商,Biopharma PEG为客户提供用于PROTAC开发的高纯度PEG连接体。我们拥有超过 3000 种高纯度 PEG 连接剂库存,可帮助您进行聚乙二醇化、生物共轭、交联、ADC 药物开发、制药和生物技术研发的生物标记。