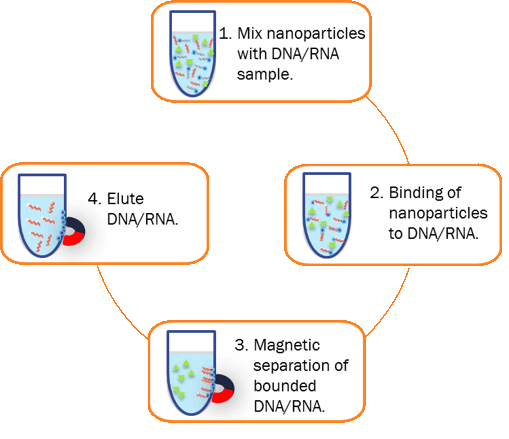
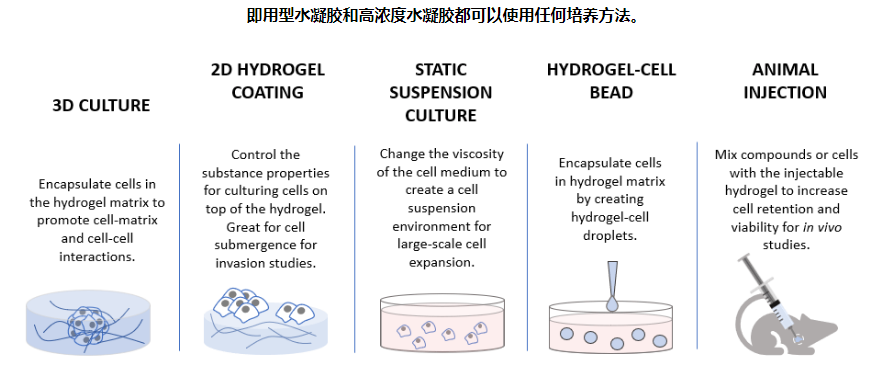
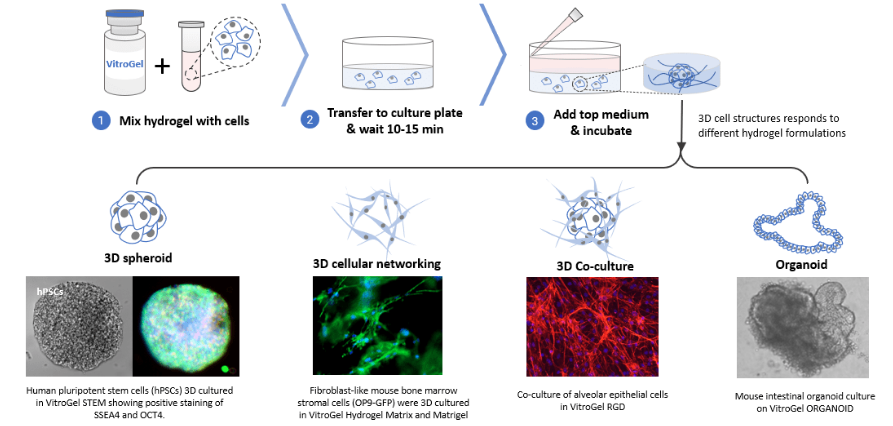
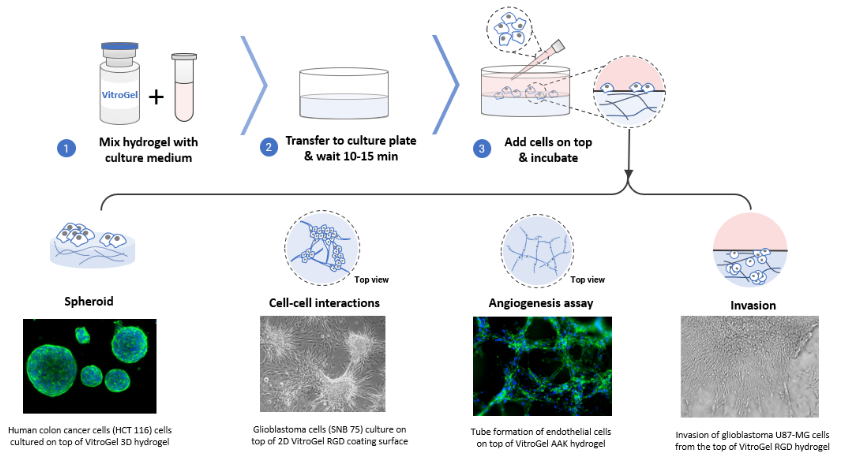
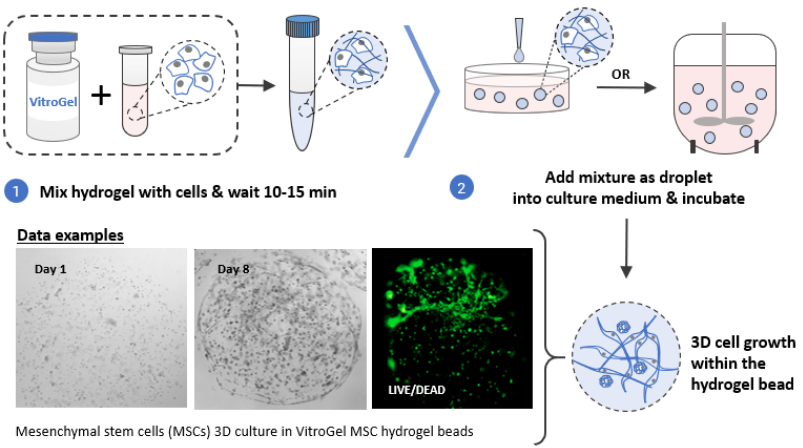
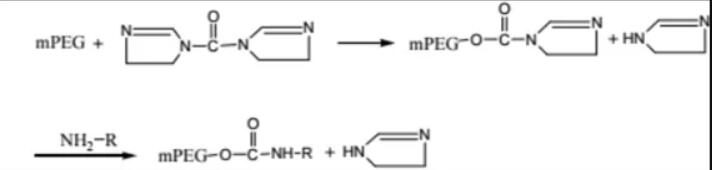
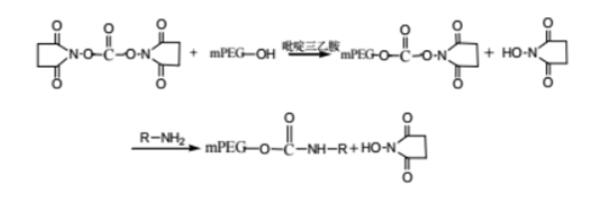
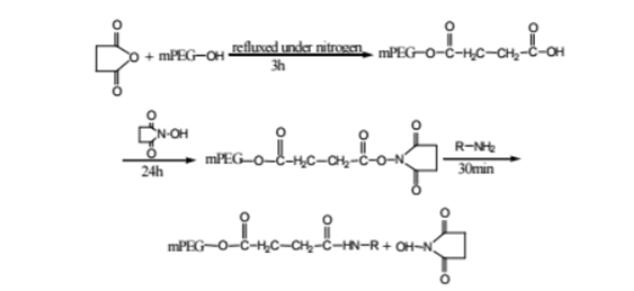
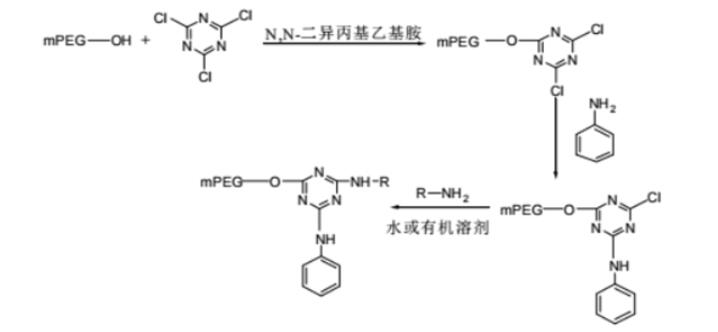
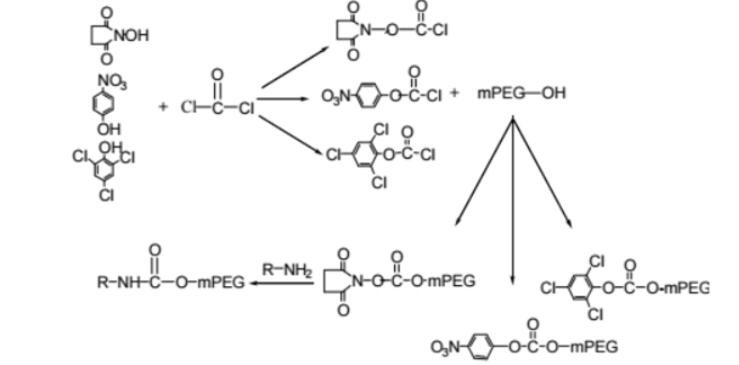
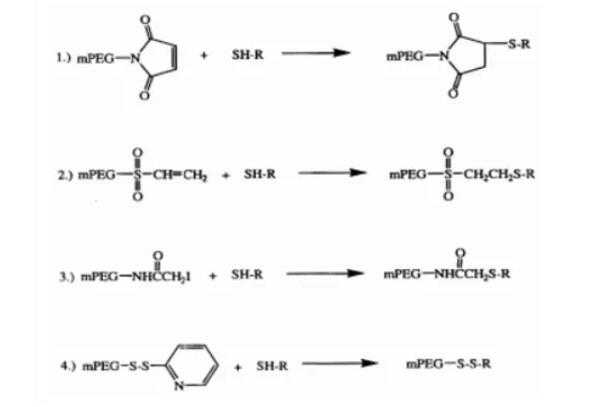
蛋白质分离纯化的方法
研究最终取决于群体的表达产物。无论是用于检测还是用于棉花保护,表达的蛋白都需要分离纯化。但蛋白质的性质不同,所以方法也不同。
蛋白质的一级、二级、三级、四级结构决定了其物理、化学、生化、物理、化学、生物性质。此外,它总结了不同蛋白质性质的差异或改变条件使它们不同。同时利用多种性质,在兼顾产量和纯度的情况下,选择蛋白质纯化的方法。
蛋白质一般以复杂混合物的形式存在于组织或细胞中,每种细胞毒性都包含数千种不同的蛋白质。蛋白质的分离和纯化是一项艰巨的任务。蛋白质纯化的总体目标是力图提高产物的纯度或比活性,要求纯化合理、快速、得率高、纯度高。并将蛋白质从细胞中的其他所有成分中分离出来,特别是不需要的不纯蛋白质,同时仍然保留肽的生物活性和化学完整性。
之所以能从数千种蛋白质混合物中纯化出一种蛋白质,是因为不同的蛋白质具有截然不同的物理、化学、理化和生物特性。
这些性质是由蛋白质的氨基酸序列和数量不同造成的,连接在多肽主链上的氨基酸残基是带正电的、带电的、极性的还是非极性的、亲水的还是疏水的。
此外,多肽还可以折叠成非常确定的二级结构(α螺旋、β折叠和各种角度)、三级结构和四级结构。利用待分离蛋白质与其他蛋白质性质的差异,在蛋白质表面形成一定大小、形状和分布的残基,并设计一套合理的分级分离步骤。
蛋白质混合物可以根据蛋白质不同性质对应的方法进行分离:
1 分子大小
不同类型的蛋白质在分子大小上有一定的差异,可以采用一些简单的方法使蛋白质混合物得到初步分离。
1.1 透析和超滤
透析在纯化中非常常用,可以去除盐(脱盐和置换缓冲液)、有机溶剂和低分子量抑制剂。透析膜的截留分子量约为5000。例如,分子量小于10000的酶溶液可能会泄漏。超滤一般用于浓缩和脱色。
1.2 更换缓冲液的离心分离
许多酶富含于某种细胞器中。匀浆后,通过离心得到某种亚细胞成分,使酶富集10-20倍,然后纯化出特定的酶。差速离心,分辨率低,仅适用于粗提取或浓缩。
速率分区,如果离心时间太长,所有物质都会沉淀。因此,需要选择最佳的分离时间,以获得相当纯的亚细胞成分进行进一步纯化,避免差速离心中大、小成分的沉淀。但容量较小,只能少量使用。
密度梯度离心常用的介质包括蔗糖、聚蔗糖、氯化铯、溴化钾和碘化钠。
1.3 凝胶过滤
这是根据分子大小分离蛋白质混合物的有效方法之一。注意待分离蛋白质的分子量在凝胶的工作范围内。选择不同分子量的凝胶可用于脱盐、更换缓冲液以及利用分子量差异去除热源。
2 形状
当蛋白质在离心过程中穿过溶液时,或者当它们穿过膜、凝胶过滤填料颗粒或电泳凝胶中的小孔时,蛋白质会受到其形状的影响。
对于相同质量的两种蛋白质,环状蛋白质的有效半径(斯托克斯半径)较小。通过溶液沉降时遇到的摩擦力很小,沉降速度更快,并且比其他形状的蛋白质显得更大。相反,在尺寸排阻色谱中,斯托克半径较小的球状蛋白质更容易扩散到凝胶过滤填料颗粒内部并随后洗脱,因此它们看起来比其他形状的蛋白质更小。
3 溶解度
利用蛋白质溶解度的差异来区分各种蛋白质的常用方法。影响蛋白质溶解度的外界因素有很多,其中主要的有:溶液pH、离子强度、介电常数和温度。但在相同的特定外部条件下,不同的蛋白质具有不同的溶解度。适当改变外部条件来控制蛋白质混合物中某种成分的溶解度。
3.1 pH控制和等电点沉淀
蛋白质在等电点通常溶解度较低。
3.2 蛋白质的盐腌和盐析
3.3 有机溶剂分类方法
蛋白质在不同溶剂中的溶解度差异很大,从基本不溶(<10μg/ml)到极易溶解(>300mg/ml)。影响蛋白质溶解度的可变因素包括温度、pH、溶剂极性、离子性质和离子强度。引起蛋白质沉淀的有机溶剂的浓度不同,因此可以控制有机溶剂的浓度来分离蛋白质。
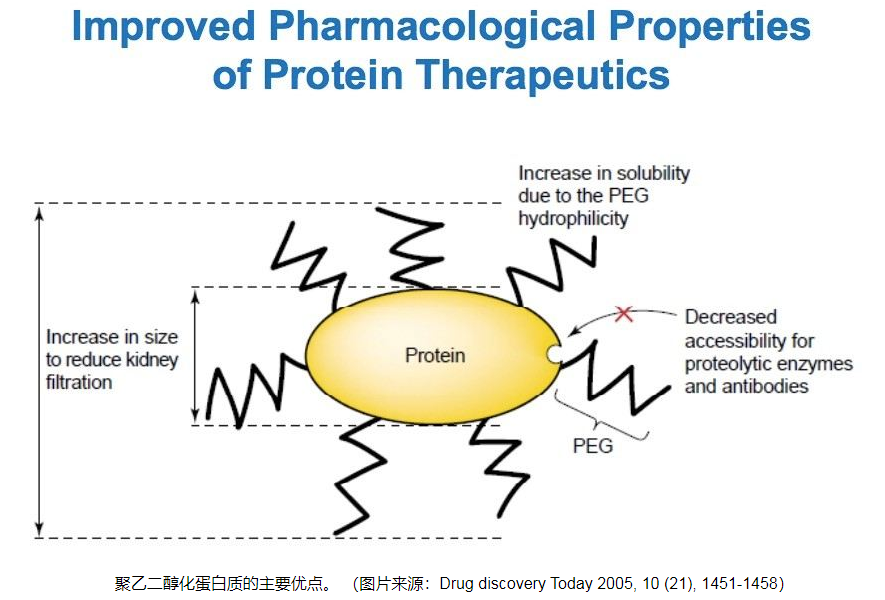
水溶性非离子聚合物(聚乙二醇)也会引起蛋白质沉淀。
3.4 温度
不同的蛋白质在不同的温度下具有不同的溶解度和活性。大多数蛋白质在低温下比较稳定,因此分离操作一般在0℃或更低的温度下进行。
4 充电
蛋白质的净电荷取决于氨基酸残基携带的正电荷和负电荷的总和。例如,带有净负电荷的中性溶液称为酸性蛋白质。
4.1 电泳
它不仅是分离蛋白质混合物和鉴定蛋白质纯度的重要方法,也是研究蛋白质性质的非常有用的方法。
等电聚焦分辨率非常高,pI可以以0.02pH的差异分离。
蛋白质 2D-PAGE 分离的分辨率已发展到 100,000 个蛋白质点。
4.2 离子交换色谱法
改变蛋白质混合溶液中的盐离子强度pH和(阴离子、阳离子)离子交换填料,不同蛋白质对不同离子交换填料的吸附能力不同,蛋白质因吸附能力不同或不被吸附而分离。
可以通过保持洗脱液组成恒定或通过改变洗脱液的盐度或pH来进行洗脱。后者又可分为分段洗脱和梯度洗脱。梯度洗脱一般效果好,分辨率高,特别是使用交换容量小、对盐浓度敏感的离子交换剂。控制洗脱液的体积(与柱床的体积相比)、盐浓度和pH,以及样品组分可以从离子交换柱中单独洗脱。
蛋白质分子外表面暴露的侧链基团的类型和数量不同,因此缓冲液在一定pH值和离子强度下的电荷也不同。
5 电荷分布
带电的氨基酸残基可以均匀分布在蛋白质表面,可以与适当强度的阳离子交换柱或与阴离子结合。由于大多数蛋白质不能在单一溶剂中与两种类型的离子交换柱结合,因此可以利用这种特性来纯化它们。带电的氨基酸残基还可以成簇分布,使得一个区域带强正电荷,另一区域带强负电荷,呈强酸性或强酸性。可与阳离子交换树脂或一定pH条件下的阳离子交换树脂结合使用。例如,钙调蛋白只能在 pH 2 时与阳离子交换树脂结合。
6 疏水性
大多数疏水性氨基酸残基隐藏在蛋白质内部,但也有一些位于表面。蛋白质表面疏水性氨基酸残基的数量和空间分布决定了蛋白质是否具有与疏水性柱填料结合用于分离的能力。
其价格低廉,纯化的蛋白质具有生物活性,是分离纯化蛋白质的通用工具。
高浓度盐水溶液中的蛋白质保留在柱上,并在低盐或水溶液中从柱上洗脱。因此,特别适合浓硫酸铵溶液沉淀分离后的母液,以及沉淀后含有目标产物的溶液用盐溶解后直接注入柱中。
7mol/盐酸胍或8mol/L尿素也适用于直接注入柱中的大肠杆菌蛋白提取物的处理,并且在复性的同时进行分离。
7 密度
大多数蛋白质的密度在1.3~1.4g/cm3之间。该特性通常不常用于蛋白质分级分离。
但含有大量磷酸盐或脂质的蛋白质的密度与普通蛋白质明显不同,因此大多数蛋白质可以通过密度梯度离心分离。
8 基因工程构建的纯化标记
通过改变cDNA,在表达蛋白的氨基端或羧基端添加少量额外的氨基酸,可以作为纯化的有效基础。
8.1 GST融合向量
待表达的蛋白与谷胱甘肽S-转移酶一起表达,然后用谷胱甘肽Sepharose 4B纯化,然后用凝血酶或因子Xa切割。
8.2 蛋白A融合载体
将待表达的蛋白与蛋白 A 的 IgG 结合位点融合在一起进行表达,并用 IgG Sepharose 进行纯化。
8.3(组氨酸标记)螯合琼脂糖凝胶
最常见的标记之一是在蛋白质的氨基末端添加6~10个组氨酸。在正常或变性条件下(8M尿素),借助其与Ni2+螯合柱紧密结合的能力,用咪唑洗涤去除或将pH降至5.9,使组氨酸全质子化,不再与Ni2+结合,使其纯化。
重组蛋白在设计和构建时已融入纯化概念。样品中大多混有破碎细胞或可溶产物。膨胀床吸附技术 STREAmLINE 适用于粗分离。
9 亲和力
兼具效率高、分离速度快的特点。配体可以是酶底物、抑制剂、辅因子和特异性抗体。
吸附后,可以改变缓冲液的离子强度和pH值来洗脱目标蛋白。也可用更高浓度的相同配体溶液或亲和力更强的配体溶液进行洗脱。
与超滤相结合,浓缩两者的优点,形成超滤亲和纯化,具有分离效率高、可大规模工业化的优点,适合初步分离。
根据配体的不同,可分为:
(1)金属螯合介质
过渡金属离子Cu2+、Zn2+、Ni 2+ 以亚胺配合物的形式结合。由于这些金属离子与色氨酸、组氨酸和半胱氨酸形成配位键,从而形成亚胺金属-蛋白质螯合物,使得含有这些氨基酸的蛋白质被该金属螯合物亲和色谱的固定相吸附。螯合物的稳定性由单个组氨酸和半胱氨酸的解离常数控制,该解离常数还受到流动相的 pH 和温度的影响。控制条件可以将不同的蛋白质彼此分离。
(2) 小配体亲和介质
配体包括精氨酸、苯甲酰胺、钙调蛋白、明胶、肝素和赖氨酸。
(3) 抗体亲和介质
免疫亲和层析,配体为重组蛋白A和重组蛋白G,但蛋白A比蛋白G特异性更强,并且蛋白G可以结合更多不同来源的IgG。
(4)颜料亲和介质
染料色谱的效果主要取决于染料配体及其与酶的亲和力大小。它还与洗脱缓冲液的类型、离子强度、pH值和待分离样品的纯度有关。有两种配体:Cibacron Blue 和 Procion Red。在某些条件下,固定化染料可以充当阳离子交换剂。为了避免这种现象,最好在离子强度小于0.1、pH大于7时进行操作。
(5) 外源凝集素亲和介质
配体包括刀豆球蛋白、扁豆凝集素和麦芽凝集素。固相凝集素可以与多种碳水化合物残基发生可逆反应,适用于多糖和糖蛋白的纯化。
10 非极性群体之间的力量
流动相中的置换剂是极性比水小的有机溶剂,这些有机溶剂可能会导致许多蛋白质分子发生不可逆的变性。流动相中必须存在离子对试剂才能使分离有效并获得高质量回收率。分离必须在酸性介质中进行,而有些蛋白质在后两种条件下会产生不可逆的分子构象变化,因此人类在生物大分子中的分离纯化受到限制。
正相色谱法在生物大分子的分离纯化中应用相对较少,因为所用的溶剂非常昂贵。
11 可逆缔合
在一定的溶液条件下,有些酶可以聚合成二聚体、四聚体等,而在另一种条件下,则形成单体。
12 稳定性
12.1 热稳定性
大多数蛋白质在加热至 95°C 时会解折叠或沉淀。利用这种特性,在加热后保留其可溶性活性的蛋白质可以很容易地与大多数其他细胞蛋白质分离。
12.2 蛋白水解的稳定性
用蛋白酶处理上清液以消化受污染的蛋白质,留下对蛋白水解具有抗性的蛋白质。
13 分配系数
采用水相两相萃取分离,常用的生物材料分离系统有:聚乙二醇(PEG)/葡聚糖(DEXTRAN)、PEG/磷酸盐、PEG/硫酸铵等。由于其含水比高,所选用的生物材料分离系统有:聚合物和盐对酶无毒。而分离设备也应用于化工行业,在工业上受到重视。
开发:新型双水相分离技术有亲和双水相萃取、膜分离双水相萃取等。
14 表面活性
14.1 泡沫分离
蛋白质溶液具有表面活性,溶液中产生气体,气泡与液相主体分离并富集在塔顶,达到分离浓缩的目的。
14.2 反胶束相转移法
反胶束相转移法是80年代出现的一种新型分离技术。它利用表面活性剂分子在有机溶剂中自发形成的反胶束。在一定条件下,水溶性蛋白质分子溶解成反胶束。在极核中,创造条件将蛋白质萃取到另一个水相,实现蛋白质的相转移,达到分离纯化蛋白质的目的。
反胶束中的蛋白质分子受到周围水分子和表面活性剂极性头的保护,仍然保持一定的活性,甚至表现出超活性。据报道,AOT/异辛烷反相胶束用于酵母脂肪酶的相转移。