PROTAC的发展概述
临床上使用的大多数药物都是基于小分子的。与传统的小分子抑制剂和拮抗剂不同,蛋白质降解技术由于能够诱导治疗靶点蛋白质的降解,近年来发展迅速,为新药的开发提供了新的思路。
PROTACs(PROteothesis-Targeting Chimeras)的概念最早由Crews等人提出。 2001年,PROTACs可以利用体内天然的蛋白质清洁系统来降低蛋白质水平而不是抑制蛋白质功能,从而治愈疾病。 PROTAC是一种异双功能分子,看起来像哑铃,分子的一端连接到结合靶蛋白的配体,一端连接到E3泛素连接酶,中间有合适的接头。 PROTAC对目标蛋白的降解是通过泛素蛋白酶体系统(UPS)实现的:PROTAC分子与目标蛋白(POI)和E3连接酶结合形成三元复合物,对目标蛋白进行泛素化标记,泛素化蛋白被泛素化酶识别并降解。细胞内蛋白酶体26S。
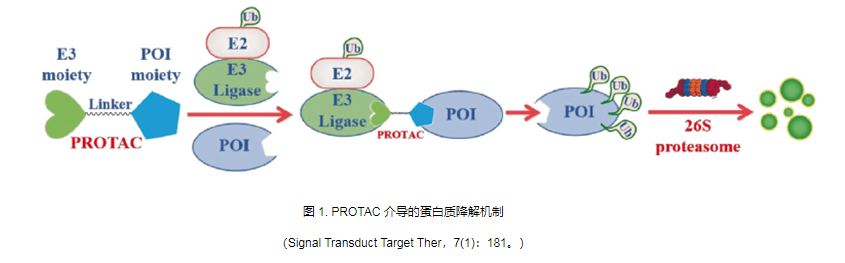
2022年6月9日,清华大学饶宇教授团队在Nature旗下Signal Transduction and Targeted Therapy杂志上发表了题为“ PROTACs:学术界和工业界的巨大机遇(2020年至2021年更新) “的综述文章,详细介绍了 近两年PROTAC技术的研究进展,总结了PROTAC针对癌症、病毒感染、免疫疾病、神经退行性疾病的代表性新靶点。
PROTACs新靶点研究进展
近20年来,PROTAC领域进入了快速发展期,特别是2015年以Pomalidomide为E3连接酶配体的dBET1 PROTAC成功降解BET蛋白以来,近两年PROTAC相关研究论文不断涌现。经历了爆发式增长。 2019年,饶宇团队总结了40多个据报道被PROTAC降解的蛋白质靶点。近两年新增了约90个可被PROTAC降解的蛋白靶点,涵盖癌症、免疫紊乱、病毒感染、神经退行性疾病等疾病领域,其中癌症是主要应用领域。
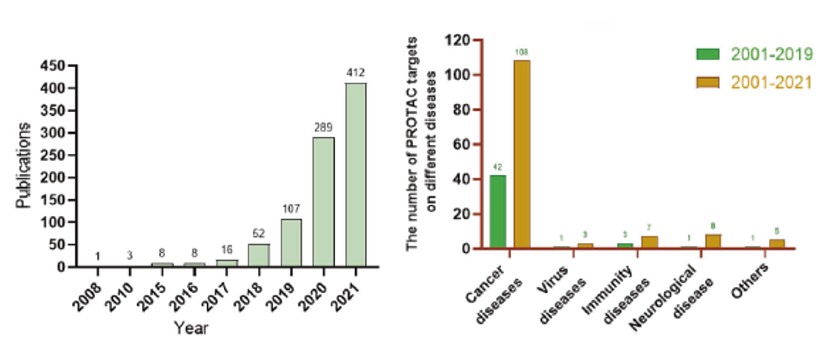
图2.近两年PROTAC相关研究的快速发展
(Signal Transduct Target Ther, 7(1): 181.)
据统计,基于PROTAC技术的不同降解剂可降解约54种激酶,占总靶点的45%。激酶已被优选作为蛋白质降解的靶标,主要是因为大多数激酶具有已知且有效的抑制剂或配体,可以轻松修饰以连接接头并保持足够的结合亲和力。此外,激酶具有深层结合袋,可促进 PROTAC 的结合,从而诱导激酶与 E3 连接酶的相互作用,进而泛素化并最终降解激酶。此外,尽管激酶蛋白具有高度同源性,PROTAC 仍可以选择性降解不同激酶亚型。
迄今为止,已发现518种激酶,参与细胞生存、增殖、分化、凋亡、代谢等多种生理调节过程。如下图所示,这些激酶根据其结构和功能可以分为九类,即受体酪氨酸激酶(RTKs)、TKL激酶(TKLs)、STE激酶(STEs)、CAMK激酶(CAMKs)、AGC激酶(AGC)、CMGC 激酶 (CMGC)、非典型蛋白激酶、CK1 激酶 (CK1) 等。其中,红色标记的是人类可降解激酶,具有现有的PROTAC降解剂。受体酪氨酸激酶(RTK)和CMGC激酶(CMGC)的PROTAC降解剂最为发达,分别有19个和14个,占总数的一半以上。相比之下,CK1 激酶 (CK1) 和 CAMK 激酶 (CAMK) 尚未开发出 PROTAC 降解剂。
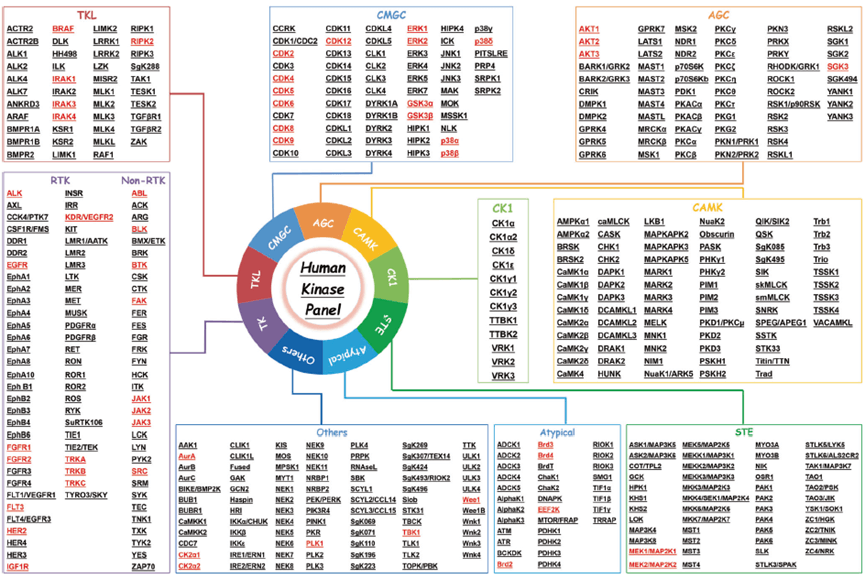
图 3. 基于 PROTAC 技术的可降解人类激酶及其分类
(Signal Transduct Target Ther, 7(1): 181.)
PROTAC新技术
由于PROTACs是在POI抑制剂的基础上发展起来的,因此仍然存在一定程度的脱靶效应。由于PROTAC的分子量较大,其细胞膜通透性差和药代动力学(PK)特性大大降低了其生物学和治疗效果。此外,有些PROTAC虽然能有效诱导靶蛋白降解,但其生物学作用较弱,对疾病没有有效作用。最后,大多数蛋白质没有相应的小分子缀合物来设计PROTAC,例如在疾病发展中发挥重要作用的转录因子。由于转录因子的抑制剂很少,因此在设计靶向转录因子的PROTAC时没有可用的粘合剂。这极大地限制了PROTAC技术的应用。 为了解决上述问题,近年来出现了不同类型的PROTAC技术,例如抗体-PROTAC、适体-PROTAC缀合物、双靶点PROTAC、叶酸笼式PROTAC和TF-PROTAC。
抗体-PROTAC
Antibody-PROTAC是一种探索新型抗体-PROTAC缀合物与抗体组装的新策略。该技术能够对不同细胞和组织中的蛋白质进行特异性降解,从而优化治疗窗口,减少广谱 PROTAC 的副作用,并增加其作为药物或化学工具的潜力。
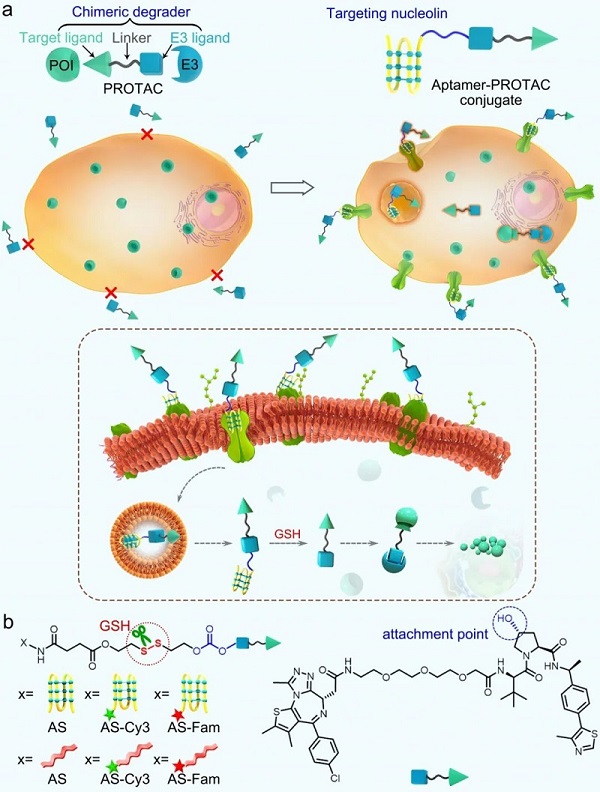
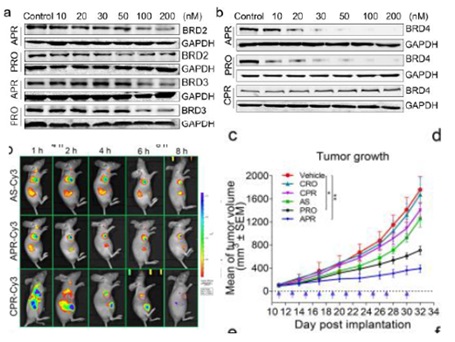
适体-PROTAC 缀合物
适配体是具有复杂三维结构的单链核酸,主要包括茎、环、发夹和G4聚合物。它们通过氢键、范德华力、碱基堆积力、静电效应等特殊作用与靶蛋白结合,具有较高的特异性和亲和力,可提高传统PROTAC的水溶性、膜通透性和肿瘤靶向性。
双目标PROTACS
在癌症的发生和发展过程中,通常有多种因素共同作用,包括不同种类的激酶和生长因子,它们可以独立作用,也可以通过信号网络相互干扰。该方法主要是设计一个结合两个或多个药效团的单一分子,同时靶向两个或多个抗肿瘤靶点。
叶酸笼式 PROTAC
叶酸受体 α (FOLR1) 在正常组织中表达较低,但在许多人类癌症中表达较高。叶酸笼式 PROTAC 是另一种提高 PROTAC 靶向特异性的技术。基本原理是将叶酸基团引入PROTAC分子中,实现在靶细胞和组织中的释放。该技术中,叶酸通过细胞内源性水解酶的作用释放出活性PROTAC,然后降解剂诱导目标蛋白的降解。
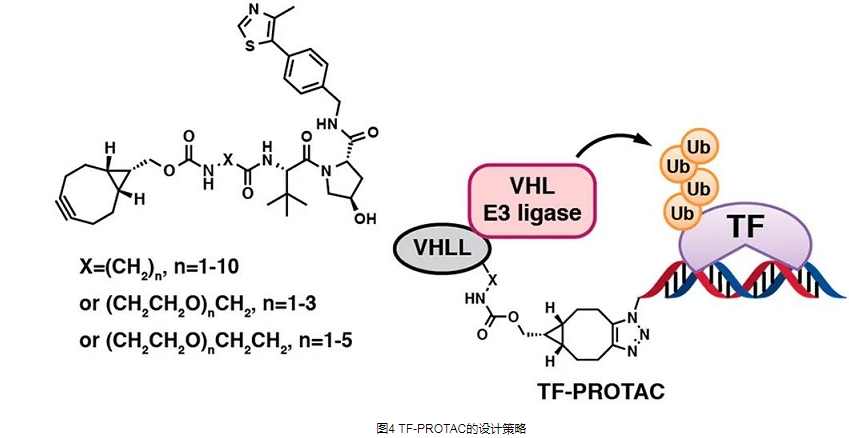
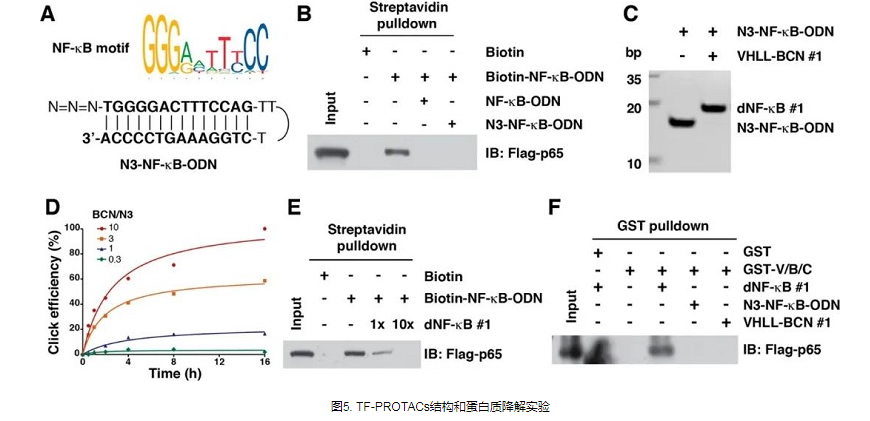
TF-PROTAC
转录因子(TF)是一类参与基因表达和调控的蛋白质,也是癌症治疗的潜在靶点。与传统激酶不同,转录因子不具有激酶或其他酶中常见的活性口袋或变构调节位点,这使得它们难以被小分子抑制剂靶向。由于TF可以结合特定的DNA序列并调节转录过程,因此理论上可以靶向具有不同DNA序列的TF而不是小分子抑制剂。因此,TF-PROTAC将靶向蛋白的小分子配体替换为相应的DNA序列,从而形成TF-PROTAC,靶向特异性TF,并诱导其降解,从而调节特异性TF的水平和生物学功能。
PROTAC的发展潜力
与其他药物和疗法相比, PROTACs具有许多潜在的优势,例如广泛的组织分布和口服给药。与其他疗法(如细胞疗法、抗体药物等)相比,PROTAC的生产工艺更为简单。与小分子药物相比,PROTAC可以靶向更多小分子药物无法靶向的靶点,从而产生更好的疗效。因此,PROTAC技术受到业界高度关注,并开始应用于癌症、免疫紊乱、病毒感染、神经退行性疾病等药物研发,其中以癌症领域的应用为主一。
PROTACs作为一项新兴且有前景的技术,在以下方面显示出巨大的发展潜力。
首先,PROTAC 对耐药靶点表现出异常的敏感性。传统上,化疗一直是癌症治疗的主要手段,但化疗药物的获得性耐药性阻碍了临床应用并导致疾病复发。后来开发出激酶抑制剂,免疫疗法也暴露了耐药性的问题。由于PROTAC通过清除整个靶蛋白来影响蛋白功能,包括酶促活性功能和非酶促功能,因此该技术有望解决当前治疗面临的潜在耐药性。
其次,PROTACs有潜力瞄准“不可成药的目标”。大多数小分子药物或大分子抗体需要与酶或受体的活性位点结合才能发挥作用;然而,据估计,人体细胞中 80% 的蛋白质都缺乏这样的位点。 PROTAC 可以通过任何角落和缝隙抓住目标蛋白质。
第三,PROTACs可以影响非酶功能。传统的小分子药物通常通过消除其靶标的酶活性来发挥作用。越来越多的研究表明,PROTACs具有扩大靶点“可成药空间”并控制蛋白酶和非酶功能的潜力。
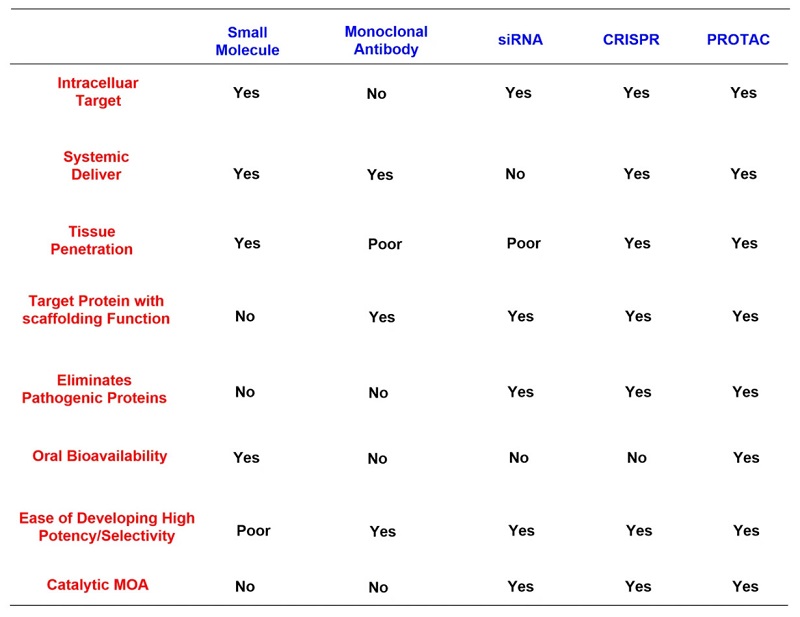
PROTAC 与其他治疗技术的比较
(来源:信号转导和靶向治疗)
Arivinas由Craig Crews创立,进行 PROTAC技术的药物开发。 近两年, Arivinas公司开发的针对雄激素受体(AR)的PROTAC抑制剂ARV-110和针对雌激素受体(ER)的PROTAC抑制剂ARV-471在前列腺癌和乳腺癌中得到了临床验证,分别是PROTAC技术应用的里程碑。
PROTACs的临床试验研究
除了作为研究工具外,PROTAC 在疾病治疗方面也具有巨大的应用潜力。它已成为一种新的药物发现模式,有潜力将传统药物发现转变为新的重磅疗法。截至2022年3月,如下表所示,全球已有十余种PROTAC药物进入临床开发阶段。其中Arvinas的ARV-110和ARV-471已进入临床II期,是PROTAC药物中临床进展最快的。
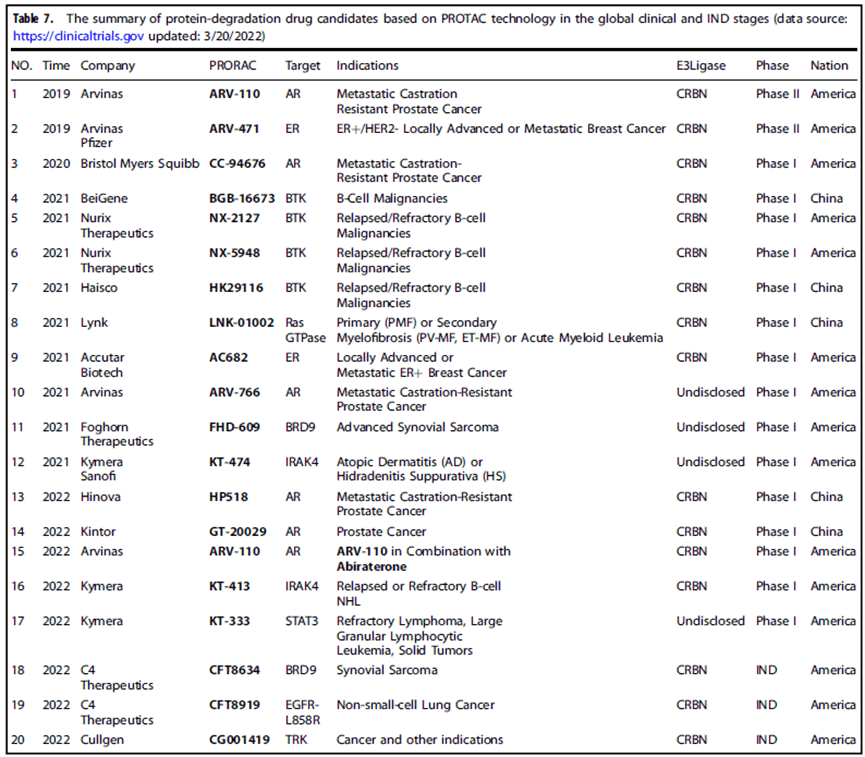
表1. 全球临床和IND阶段基于PROTAC技术的蛋白降解候选药物汇总
(Signal Transduct Target Ther, 7(1): 181.)
结论
与其他药物和疗法相比,PROTACs具有许多潜在的优势,例如广泛的组织分布和口服给药。与其他疗法(如细胞疗法、抗体药物等)相比,PROTAC的生产工艺更为简单。与小分子药物相比,PROTAC可以靶向更多小分子药物无法靶向的靶点,从而产生更好的疗效。因此,PROTAC技术受到业界高度关注,并开始应用于癌症、免疫紊乱、病毒感染、神经退行性疾病等药物研发,其中以癌症领域的应用为主一。由克雷格·克鲁斯 (Craig Crews) 创立的阿里维纳斯 (Arivinas) 是第一个推出该药物的公司PROTAC技术的发展。近两年,Arivinas公司开发的针对雄激素受体(AR)的PROTAC抑制剂ARV-110和针对雌激素受体(ER)的PROTAC抑制剂ARV-471在前列腺癌和乳腺癌中得到了临床验证,分别是PROTAC技术应用的里程碑。
尽管PROTAC在过去20年中发展迅速,但仍有许多挑战需要解决。这些挑战主要来自两个方面,即PROTAC分子设计和成药性的优化,以及生物活性的综合评价。第一个是关于 PROTAC 的分子设计和成药性,涉及靶蛋白配体、新的 E3 连接酶配体和新的接头。二是生物活性评价,涉及PROTAC分子的筛选、成药性评价和药理评价。这些问题目前还没有现成的答案,但相信随着更多生物学、药理学和临床研究的发展,新的评价方法和体系将逐步建立来解决这些问题。相信未来会有越来越多的PROTAC进入临床前和临床研究,这将进一步测试PROTACs的治疗效果。预计未来PROTAC技术将为人类疾病治疗和生命健康带来裨益。
PROTAC 开发中常用的接头是 PEG。Biopharma PEG是一家专业的PEG衍生物供应商,提供多功能PEG衍生物作为PROTAC连接体。我们拥有 3000 多种高纯度PEG 连接体库存,为客户的制药和生物技术研发领域的聚乙二醇化、生物共轭、ADC 药物开发提供支持。